Transgenes typically have all, or all but the first, intron removed. Still, splicing at cryptic splice sites occurs. This is linked to remnant exonic splice enhancer motifs. Removing these motifs alongside with other unwanted functionality will thus improve transgene efficiency.
How it works
Generating variants and selecting the best
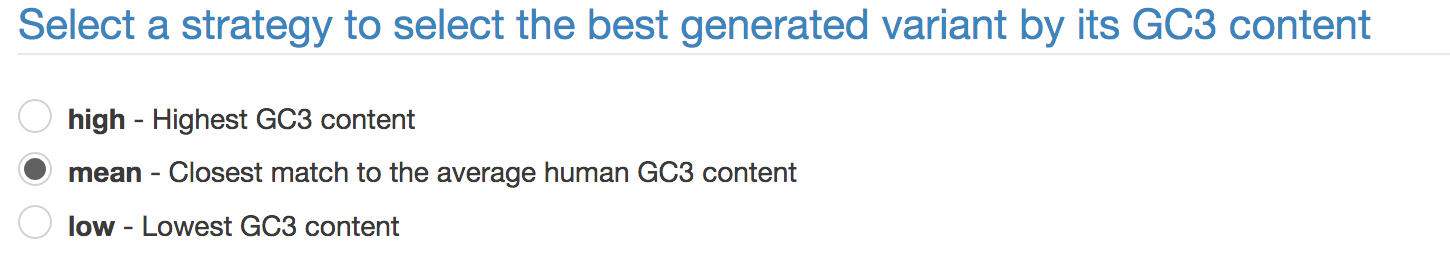
The SequenceOptimizer software will generate 1,000 gene variants and select the best by its GC3 content. By default, target GC3 content will be the average GC3 in human one- or two-exon genes (option mean). Alternatively, you may set target GC3 to high or low selecting for highest or lowest GC3 found among generated variants.
Removing introns determines which sites are checked for ESE resemblance
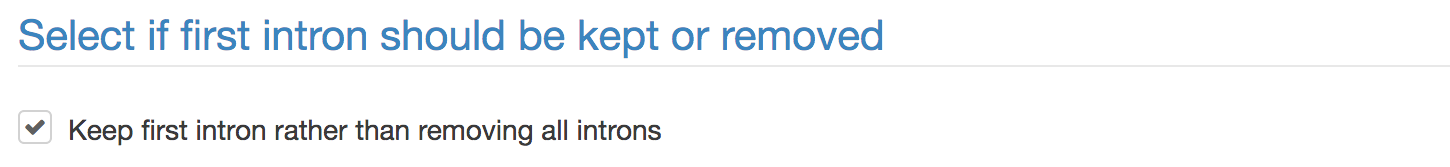
The first step when generating the gene variants is to remove all introns but the first, or, if requested all introns including the first. This is to determine exonic sites in vicinity to now deleted introns. ESE resemblance will be adjusted at those sites only and at no other position in the gene.
Per default ESE motifs will be depleted (option deplete), select option enrich to enrich them instead.
Scoring synonymous codons
For each site, synonymous codons are assigned a score and selected with a probability equaling its score. Scores are assigned based on how well the codon matches human codon usage (option humanize; default), alternative strategies include maximizing GC3 content (option max-gc) and matching the position-dependent GC3 content of human one- or two-exon genes (option gc). Should ESE motifs have been provided, a strategy to score by ESE resemblance only may also be chosen (please note: this will affect only sites near deleted introns, at all other sites the sequence will remain unchanged; option raw).
At sites in vicinity to deleted introns the codon score is a mixture of strategy-score and ESE resemblance-score. You may chose to adjust ESE resemblance at all sites instead of only at sites near deleted introns. This is not recommended as it is against our current understanding of ESEs, but may prove useful at times, e.g. when tweaking natural one-exon genes.
Synonymous codons at 6-fold degenerate sites
At 6-fold degenerate sites (leucine, serine or alanine positions) all six synonymous codons are scored per default. You can specify to restrict codons to those of the respective 2- or 4-codon sub-box instead.
Dealing with restriction sites
To preserve restriction sites already present in the sequence, please provide the corresponding recognition sequence(s) in the keep intact input tab. The SequenceOptimizer software will leave those sites intact when tweaking the gene.
Similarly, you may specify recognition sequences that are to be avoided. Please note: this will not remove restriction sites that are already present in the gene.
You may provide sites to keep intact and sites to avoid both combined or individually.